
The use of real-world evidence (RWE) has proven pivotal to research in infectious diseases. RWE’s influence is particularly evident in light of COVID-19. Amy Abernethy, M.D., Ph.D., Principal Deputy Commissioner at the U.S. Food and Drug Administration (FDA), recently shared that the agency is “approaching the generation of RWE for COVID-19 with a sense of urgency.”
RWE provides real-time insights to understand the disease landscape, to identify high-risk subgroups and potential therapies, and to establish the infrastructure for assessing the safety and tolerability of novel vaccines. In a 2005 guidance, the FDA wrote that “postmarketing safety data collection and risk assessment based on observational data are critical for evaluating and characterizing a product’s risk profile and for making informed decisions on risk minimization.” In 2019, RWE played a role in 37 percent of novel drug and biological product approvals (Source: Aetion analysis; FDA approval documents).* For therapeutics and prophylactics that target infectious diseases, seven of eight approvals included an RWE study. Of those eight approvals, FDA approved 63 percent, or five of eight, with the help of RWE.
In this article, we examine four approvals for products intended to treat or prevent infectious diseases from 2019 that included RWE studies: DENGVAXIA® (Dengue Tetravalent Vaccine, Live), EGATEN™ (triclabendazole), JYNNEOS® (Smallpox and Monkeypox Vaccine, Live, Non-Replicating), and RECARBRIO™ (imipenem, cilastatin, and relebactam). These cases highlight how foreign postmarketing data improves our understanding of a product’s risk-benefit profile.
As stated previously, of the eight therapeutics and prophylactics that the FDA approved in 2019 to target infectious diseases, seven approvals included RWE studies. Below are salient characteristics of all eight approvals related to infectious diseases.
Overview of all FDA approvals in infectious diseases from 2019:
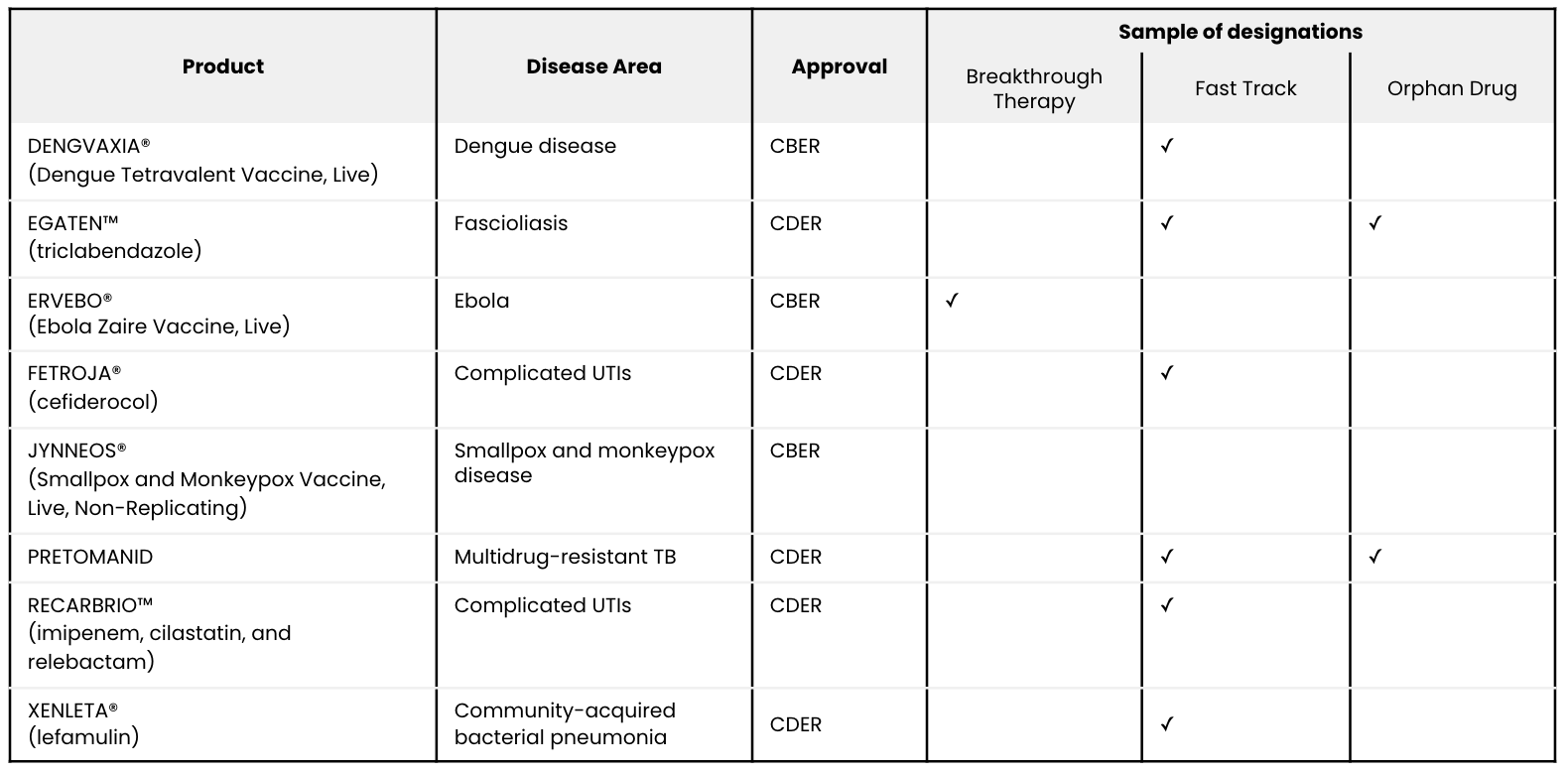
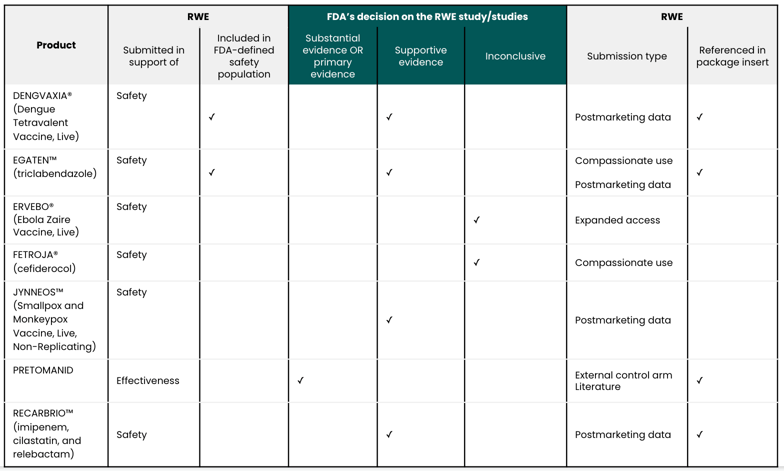
Among these approvals, seven included an RWE study:
EXAMPLE 1: DENGVAXIA indicated to prevent dengue disease associated with dengue virus subtypes 1, 2, 3, and 4
On May 1, 2019, the FDA approved Sanofi Pasteur’s DENGVAXIA (Dengue Tetravalent Vaccine, Live) to prevent dengue in individuals aged nine to 16 in patients with evidence of prior infection living in endemic areas.
Key findings from the FDA’s Summary Basis for Regulatory Action and the Pharmacovigilance Plan Assessment:
The FDA Review Committee recommended approval of DENGVAXIA based on their assessment of the applicant’s pre-clinical, clinical, and product-related data.
The Biologics License Application (BLA) included an RWE study involving postmarketing data, which contributed to the FDA Review Committee’s recommendation. The applicant obtained the first marketing authorization for DENGVAXIA in Mexico on December 8, 2015. Since then, 21 countries have approved DENGVAXIA. The majority of DENGVAXIA doses were distributed in the Philippines and Brazil, from its initial marketing authorization to August 31, 2018. In the Philippines, at least 835,000 individuals received one or more doses during public immunization programs; in Brazil, approximately 300,000 individuals received doses.
As of September 14, 2018, Sanofi Pasteur received reports of 487 serious adverse events (SAEs) and 2,283 non-SAEs. In the package insert, the FDA required Sanofi Pasteur to list disorders of the immune system and infections and infestations as potential adverse events while a patient is taking DENGVAXIA. The FDA explains that “because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to the vaccine.”
EXAMPLE 2: EGATEN indicated to treat fascioliasis
On February 13, 2019, the FDA approved Novartis’ EGATEN (triclabendazole) “for the treatment of fascioliasis in patients 6 years of age and older.”
CIBA-Geigy, Novartis’s predecessor, developed triclabendazole; it was introduced for veterinary use as Fasinex in 1983, then for human use in 1986.
Key findings from the FDA’s Multi-Discipline Review and Other Review(s):
Supportive evidence of safety includes compassionate use program data and postmarketing data.
As seen in the postmarketing data, between 2006 and 2018, Novartis donated an estimated 2.6 million treatment courses. The applicant submitted the Periodic Safety Update Report from the Novartis Global Safety Database based on the most recent review period: November 2012 through March 2017. The report did not demonstrate new or significant safety signals. However, it did note a signal for drug resistance, which was added to the Core Data Sheet.
EXAMPLE 3: JYNNEOS indicated to prevent smallpox and monkeypox disease
On September 24, 2019, the FDA approved Bavarian Nordic’s JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for adults aged 18 years or older, to prevent smallpox and monkeypox infection. Previously, in 2013, the European Medicines Agency (EMA) approved the product under the trade name IMVANEX®, and the Public Health Agency of Canada approved it under the trade name IMVAMUNE®.
Key findings from the FDA’s Summary Basis for Regulatory Action and the Pharmacovigilance Original BLA Memo:
In addition to the clinical trials, postmarketing data influenced the FDA’s determination of product safety.
There is a risk of myopericarditis in individuals who receive ACAM200, a smallpox vaccine. Therefore, the applicant monitored for cardiac adverse events of special interest (AESI). There was only one possible case of acute pericarditis in the clinical trials and no reported cases of myopericarditis in the postmarketing database, which included approximately 4,000 subjects. In the Pharmacovigilance Original BLA Memo, the FDA deemed that “the combination of routine pharmacovigilance, enhanced surveillance in the form of event-specific questionnaires, the mass vaccination study, and labeling of cardiac AESI from the clinical studies is adequate for monitoring the potential risk of inflammatory cardiac disorders.”
EXAMPLE 4: RECARBRIO indicated to treat complicated urinary tract infections (UTIs)
On July 16, 2019, the FDA approved Merck’s RECARBRIO (imipenem, cilastatin, and relebactam) to treat complicated UTIs. At the time of the submission, the combination therapy—referred to as IMI/REL—had not yet been marketed. However, imipenem/cilastatin, which is a component of IMI/REL, was approved in the U.S. in 1985 for various bacterial infections, including UTIs.
Key findings from the FDA’s Multi-Discipline Review:
The FDA’s assessment of efficacy relied to some extent on previous findings for imipenem—in vitro and animal data—and the FDA’s own analyses on the probability of target attainment for relebactam. The FDA’s assessment of safety relied on 10 trials and postmarketing data with imipenem/cilastatin.
During a pre-New Drug Application (NDA) meeting in 2018, the FDA requested a safety database of 300 subjects receiving the proposed dose and duration of the combination therapy. While the safety database only included 269 subjects who received the proposed dose and duration, the FDA considered the database sufficient, particularly in light of the clinical experience with imipenem/cilastatin. The adverse events identified in the trials were generally consistent with prior knowledge on imipenem/cilastatin.
Adverse reactions to imipenem/cilastatin that were identified in the postmarketing setting included gastrointestinal, hematologic, central nervous system, special senses, skin, body as a whole, and renal.
Based on the clinical studies and the postmarketing data, the FDA required that the package insert include adverse reactions to imipenem/cilastatin. Broad categories include: blood and lymphatic disorders, nervous system disorders, hepatobiliary disorders, skin and subcutaneous tissue disorders, and laboratory investigations.
Note that on June 5, 2020, the applicant received FDA approval for a supplemental indication: hospital-acquired and ventilator-associated bacterial pneumonia. While this expansion was based on a randomized controlled trial, RWE will be needed in the postmarket setting.
Industry-wide shared learning process
These four cases illustrate the use of RWE for label expansion and postmarketing safety reporting. While each application is context specific, sponsors should understand precedent law and other variables that will affect its strategy and decision to use an RWE study. FDA Commissioner Stephen Hahn, M.D., stated, “The more experience we have with real-world evidence, the more confidence we will have in using it for product decisions.” As demonstrated in the four examples, real-world data, when fit for purpose, has been used to generate evidence to support product approval.
As Aetion systematically assesses approval documents for drugs and biologics, we will highlight best practices on how to approach RWE. We invite you to learn along with us about the FDA’s decisions.
* We excluded assays, blood grouping reagents, and solutions from our analysis (n=21).